
Introduction
In the complex landscape of drug development, Investigational New Drug (IND) maintenance plays a pivotal role in ensuring the safety of participants enrolled in the studies, upholding data integrity, and fostering efficient communication with regulatory authorities. This article delves into the critical aspects of IND maintenance, elucidating its significance in the drug regulatory affairs landscape.
Importance of IND Maintenance
Patient Safety:
IND maintenance encompasses the submission of safety reports, a crucial component that allows the FDA to monitor the investigational drug’s safety in real-time. Swift identification of potential risks and adverse effects is paramount for protecting the well-being of trial participants.
Data Integrity:
Regular updates to the FDA through IND maintenance include but are not limited to information amendments and protocol amendments. These updates are subject to FDA comments, which ensure the integrity of clinical trial data by promptly reflecting any changes in the investigational plan.
Progress of Drug Development:
Sponsors are required to submit a brief report on the progress of the investigation within 60 days of the anniversary date of the IND. This periodic reporting aids the FDA in monitoring the drug development process, contributing to informed decision-making during the product marketing application stage.
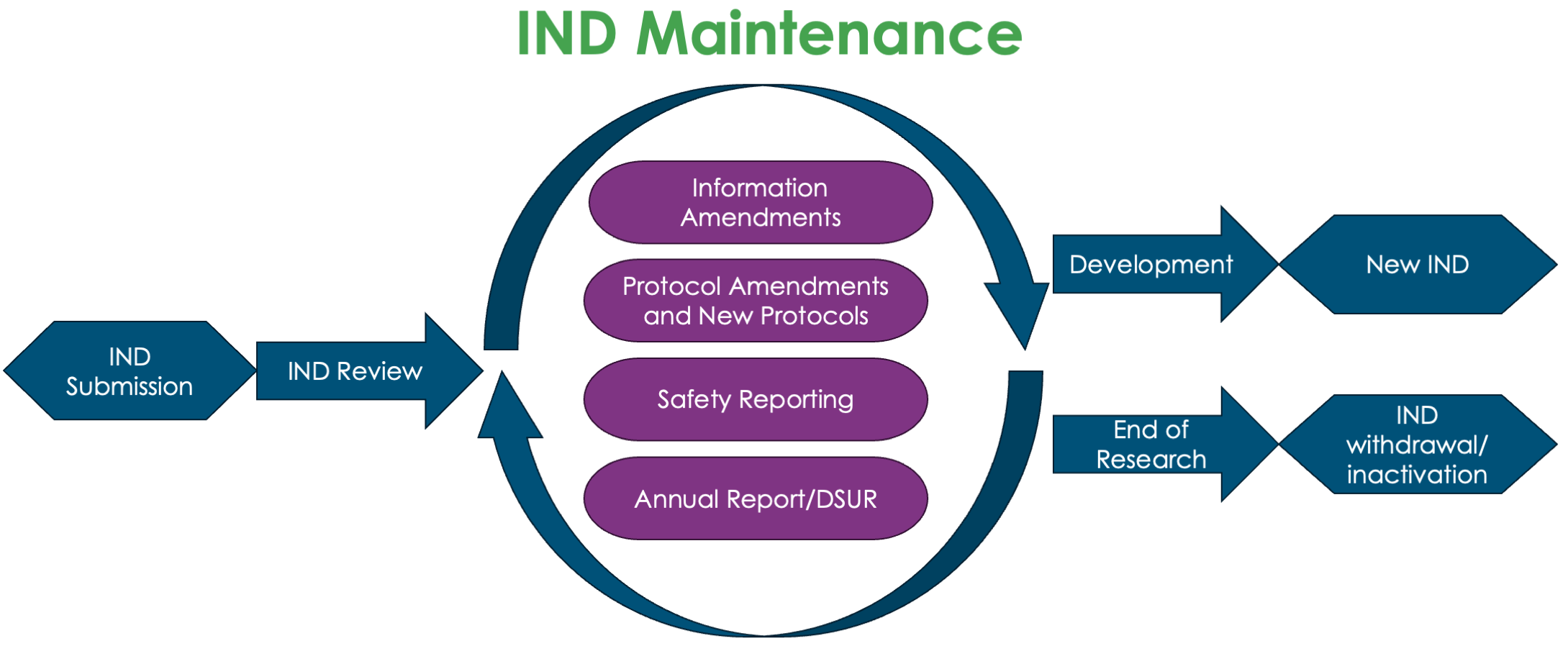
Figure 1: The role of IND maintenance in IND lifecycle
Failure to adequately maintain an IND can lead to the FDA placing the IND on clinical hold, disrupting the progress of the clinical trial. In essence, IND maintenance is more than a regulatory requirement; it is a sponsor’s commitment to monitoring patient safety, maintaining data integrity, and facilitating transparent communication with regulatory authorities.
Safety Reporting for INDs
Regulations and guidance:
21 CFR 312.32: eCFR :: 21 CFR 312.32 — IND safety reporting.
The regulation provides requirements related to IND safety reporting, specifying the requirement that a Sponsor must report any suspected adverse reaction that is both serious and unexpected indicating potential risks related to the study drug, the safety information could be derived from clinical trial or any other sources (including but not limited to findings from other studies, animal or in vitro testing). “suspected adverse reaction” means adverse event for which there is a reasonable possibility that the drug caused the adverse event.
FDA Guidance for Industry: Safety Reporting Requirements for INDs and BA/BE Studies (fda.gov)
Before submitting an IND safety report, the sponsor must ensure that the event meets the criteria for a suspected unexpected serious adverse reaction (SUSAR). The timing of submission varies, with events categorized as 7-day or 15-day reporting based on their nature and severity.
SUSAR reporting timeline:
7-day Reporting
- Events that are unexpected and fatal or life-threatening require rapid reporting to the FDA.
15-day Reporting
- Serious and unexpected suspected adverse reactions (21 CFR 312.32(c)(1)(i))
- Findings from other studies that suggest a significant risk in humans exposed to the drug (21 CFR 312.32(c)(1)(ii))
- Findings from animal and in-vitro testing that suggest a significant risk in humans exposed to the drug (21 CFR 312.32(c)(1)(iii))
- Increased rate of occurrence of serious suspected adverse reactions (21 CFR 312.32(c)(1)(iv)), that, is of adverse reactions already thought to be drug-related.
- Serious adverse events from bioavailability and bioequivalence studies not under IND (21 CFR 320.31)
Discussions about safety reporting:
- Unblinding for the SUSAR reports: As discussed in the FDA Guidance for Industry, Safety Reporting Requirements for INDs and BA/BE Studies, the blind should ordinarily be broken for SUSAR reports submitted to FDA and all participating investigators. Knowledge of the treatment received is necessary for interpreting the event, may be essential for the medical management of the subject, and may provide critical safety information about a drug that could have implications for the ongoing conduct of the trial (e.g., monitoring, informed consent). The Agency does not believe that unblinding single or small numbers of serious and unexpected adverse event cases will compromise the integrity of the study, in part because such unblinding should be infrequent.
- Cross reporting of SUSAR that occurred in study sites outside of US: per 21 CFR 312.32(c)(1)(ii) The sponsor must report any findings from the clinical studies of the study drug, whether or not conducted under an IND, and whether or not conducted by the sponsor, that suggest a significant risk in humans exposed to the study drug. Ordinarily, such a finding would result in a safety-related change in the protocol, informed consent, investigator brochure (excluding routine updates of these documents), or other aspects of the overall conduct of the clinical investigation.
Annual Report
Regulation:
21 CFR 312.33 Annual Reports:
Sponsors must submit an annual report within 60 days of the IND’s anniversary date, summarizing the progress of the investigation. The update includes clinical, nonclinical and CMC updates of the study product, the general investigational plan of the study, and the update on foreign marketing development etc.
DSUR (Development Safety Update Report): International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) E2F guidance
To promote International harmonization, FDA has accepted the DSUR to meet an IND application annual report requirements and adopted the E2F guidance, which describes a common standard for periodic reporting on drugs under development among the ICH regions.
ELIQUENT Insight: Both the IND annual report and DSUR provide essential information to regulatory authorities, the IND Annual Report offers a comprehensive overview of various aspects of the clinical trial, while the DSUR places a specific emphasis on the safety profile of the investigational drug. The potential integration of the DSUR as a replacement for the IND Annual Report highlights FDA’s commitment to global harmonization and an increased focus on safety reporting in clinical trials.
Common questions about DSUR:
- DIBD (Development International Birth Date): the DSUR reporting period starts from the DIBD, which is the sponsor’s first authorization to conduct a clinical trial globally.
- Harmonization Request: If the DIBD and IND Study May Proceed date are different, a harmonization request could be submitted to FDA.
Regulatory Updates about IND Maintenance
DSUR to Replace IND Annual Report:
On December 9, 2022, The FDA was proposing to replace the current annual reporting requirement for INDs with the more comprehensive FDA DSUR format. The primary objective is evidently to gather as much of safety data as possible in an earlier stage of drug development. Given the increasing complexity of clinical studies, the FDA anticipates DSUR will present more comprehensive data. This could potentially enable the identification of specific safety signals that might otherwise remain undetected until later stages of the study. The Agency emphasizes that aligning the DSUR with the format and content supported by the ICH serves the purpose of enhancing efficiency in the annual reporting process for sponsors. By adopting a uniform format, sponsors can streamline submissions not only to the FDA but also to multiple regulatory authorities in global regions. This approach aligns with the FDA’s overarching goal of promoting international harmonization of regulatory requirements to the extent that it is both appropriate and feasible.
SUSAR Submission (FEARS System):
FDA is taking steps toward requiring electronic submission in E2B(R3) format of certain individual case safety reports (ICSRs) for products being evaluated by the FDA under an investigational new drug (IND) application with the FDA’s Adverse Event Reporting System (FEARS system). This process change would allow the FDA to access and review both pre- and post-market safety information in the same system and with the same data standard. Eliminating the need to submit the Safety Reports in eCTD package format, which means no further cover letter and Form 1571 would be needed for most SUSAR reporting.
However, FDA is not currently implementing the change, the Sponsor would still need to continue submitting IND safety reports using eCTD format and IND-exempt BA/BE safety reports on Form FDA 3500A. FDA will update their web page when final guidance for IND safety reporting is published, and when FDA will accept IND and IND-exempt BA/BE safety reports in E2B(R3) format on a voluntary basis and when it is required for all the Sponsors.
Conclusion
In conclusion, IND maintenance is a multifaceted commitment, encompassing safety reporting, data integrity, and transparent communication. Understanding and adhering to the regulatory requirements outlined in this article are foundational steps to navigate the intricate landscape of drug development successfully. Through diligent IND maintenance, sponsors contribute to the collective goal of advancing safe and effective therapies for the benefit of patients worldwide.
Author Bio:
Ziqi Zhang, Clinical Consultant / Safety Specialist. Ms. Zhang graduated from Fudan University with Bachelor of Medical Science (Preventive medicine), Duke University with Master of Global Health. Since joining ELIQUENT Life Sciences (Legacy DataRevive), Ms. Zhang mainly manages IND maintenance service, which includes safety reporting, clinical protocol/IB amendment; DSUR and ODD annual report. She manages maintenance for projects in Phase I, II, III and post marketing stage. Product modalities include small molecules, monoclonal antibodies, bispecific antibodies, ADC product, gene and cell therapy products, targeting different indications involving solid tumors, autoimmune diseases, and central nervous system (CNS) diseases.

PDF format of this white paper