
Control of Nitrosamine Impurities in Human Drugs
Guidance for Industry (Revision 2, Sep 2024)
The risk of nitrosamine impurities came to the forefront following the identification of nitrosodimethylamine (NDMA) in the angiotensin II receptor blocker, valsartan, in June 2018. After studying the issue, the FDA published a guidance in Sep 2020 regarding the Agency’s initial thoughts about some small molecule nitrosamines. Most recently in September 2024, the FDA issued a revised guidance which explains the Agency’s updated thinking about how drug manufacturers and applicants can detect and prevent unacceptable levels of nitrosamine impurities in their products. They approached this guidance by classifying nitrosamine impurities into two subsections: small molecule nitrosamines and nitrosamine drug substance-related impurities (NDSRIs). The recommendations made in this guidance apply to chemically synthesized API, DP (including biological products) containing chemical synthesized APIs, semisynthetic and fermentation products that are at risk due to their structures.
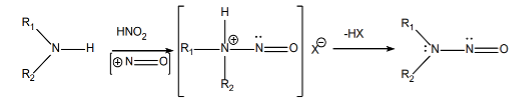
The formation of these two types of nitrosamine impurities — small-molecule nitrosamine impurities and nitrosamine drug substance-related impurities (NDSRIs) — are shown below:
Formation of Small-Molecule Nitrosamines
Formation of NDSRIs
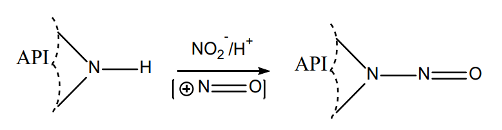
The formation of nitrosamines requires
- Presence of amines (secondary, tertiary, quaternary), and
- Presence of nitrite salts or nitrous acid, and
- Acidic conditions
In this guidance, the FDA outlines an approach for assessing the risk of nitrosamine impurities and establishing appropriate limits.
Root Causes of Nitrosamine Impurities
Nitrosamines may form in the presence of secondary, tertiary, quaternary amines and nitrosating reagents like nitrite salts and nitrous acid. Nitrosamine impurities found in API may come from materials used during the manufacturing process, including:
- API or API degradants
- Starting materials and impurities
- Intermediates
- Reagents/solvents/catalysts and impurities/degradants
- Recovered reagents/solvents/catalysts
- Quenching process (for example, nitrous acid is commonly used to decompose residual azide)
- Lack of process optimization and control
NDSRIs carried over from APIs is another source of nitrosamine impurities. Other potential causes of nitrosamine impurities found in drug products include:
- Nitrite impurities found in excipients
- Nitrite impurities in potable water
- Container closure systems
- NDSRI levels may increase during storage
Establishment of Acceptable Intake Limits of Nitrosamine Impurities
Per ICH M7 (R2), N-nitroso compounds are considered a cohort of concern which are theoretically associated with a potential for significant carcinogenic risk at intakes below the threshold of toxicological concern (1.5 µg/kg per day). Therefore, alternative methods for establishing levels of acceptable intake (AI) limits of nitrosamine impurities need to be used. The following approaches are recommended by the guidance:
- AI limits for certain NDSRIs and small-molecule nitrosamines on the nitrosamine guidance web page (https://www.fda.gov/regulatory-information/search-fda-guidance-documents/cder-nitrosamine-impurity-acceptable-intake-limits).
- The carcinogenic potency categorization approach (CPCA) detailed in Recommended Acceptable Intake Limits for Nitrosamine Drug Substance-Related Impurities (NDSRIs) (August 2023) (RAIL guidance)
- Database and literature searches for available carcinogenicity and bacterial mutagenicity data or in vivo and/or in vitro testing using the specific compound (see Appendix B in this guidance)
- If the AI limit cannot be determined using the approaches described above, FDA recommends that 26.5 ng/day be used as the AI limit.
If there is more than one nitrosamine impurity:
- The total limit of nitrosamine impurities should not exceed the recommended AI limit for the most potent nitrosamine in the drug product, or
- The total nitrosamine impurities should be controlled using the flexible AI limit approach detailed in Appendix C. In this approach, the sum of the percent of each nitrosamine level (in ppm) relative to the corresponding acceptance criterion (in ppm) is calculated. If the sum is more than 100%, the total nitrosamine level is not acceptable.
Strategy to Address the Risk of Nitrosamine Impurities
API and drug product manufacturers and applicants should use the following three-step strategy to mitigate nitrosamine impurities (both small-molecule nitrosamines and NDSRIs) in their products. It is expected by the Agency that all companies finish the confirmatory testing and submit a plan to the Agency regarding how to mitigate the risk of nitrosamine impurities by Aug 1, 2025.

Implementation of Recommended AI Limits
According to the three-step strategy, if the risk assessment (step 1) indicates that a drug product is at risk of nitrosamine formation, manufacturers and applicants should perform confirmatory testing to determine whether nitrosamines are present.
- If the confirmatory testing indicates that nitrosamine levels are not more than 10 percent of the recommended AI limit, then a release/stability specification for nitrosamine is not needed.
- If the confirmatory testing indicates nitrosamine levels exceed 10 percent of the recommended AI limit but are within the recommended AI limit, a control for nitrosamines should be established in the release and stability specifications. This change should be reported as a supplement as changes being effected in 30 days (CBE 30).
- If the confirmatory testing indicates nitrosamine levels exceed the recommended AI limit and changes in formulation, manufacturing process, or packaging are warranted, manufacturers and applicants should implement such changes that are demonstrated to ensure that nitrosamine levels remain within the recommended AI limit. This change should be reported as a supplement as prior approval supplement (PAS).
Requirements for Bioequivalence (BE) or Bioavailability (BA) Study
To reduce or prevent nitrosamine impurities, the product may need to be reformulated. In this guidance, the FDA provided two reformulation approaches which might inhibit the formation of nitrosamine impurities.
- Addition of antioxidants (ascorbic acid, α-tocopherol, propyl gallate, or cysteine hydrochloride)
- Addition of pH modifiers to modify the microenvironment to neutral or basic pH in the drug product formulation
Generally, adding a new excipient to an approved drug is considered a Level 3 change, which normally requires in vivo BE/BA studies in addition to chemistry and dissolution documentation.
The FDA recognizes that a large number of approved drug products may contain APIs at risk for nitrosamine formation. The challenge, then, is to balance the need to address nitrosamine impurities while also maintaining drug supply and minimizing market disruptions.
Based on the results of studies initiated by FDA, the addition of small amounts of certain antioxidants to biopharmaceutics classification system (BCS) III drugs does not affect the permeability of the drug substance or intestinal transporter activities. Therefore, it would not be expected to change drug absorption in vivo or affect BA or BE.
The guidance provides the FDA’s thoughts for how a company with an approved product could demonstrate BE/BA for a product requiring reformulation (Table 1).
Table 1 Demonstrating BE/BA in Reformulated Drug Product

How Eliquent Life Sciences Can Help
Risk of nitrosamine in drugs is now a global problem across pharmaceutical industry. Time is very limited for API and drug product manufacturers to address the issue. Our professional team at Eliquent Life Sciences, consisting of experts with extensive industry development experience and FDA review experience, can provide clients with appropriate advice to mitigate the nitrosamine risk.
Because we know what FDA expects, we can effectively assist clients by ensuring that supplements/amendments needed to address the risk of nitrosamine are written in a clear, accurate and logical way, with good English grammar and formatting to help avoid confusing the regulatory reviewers.
For more information visit Drugs & Biologics – Eliquent
Ready for regulatory clarity? Contact Us.
(* Indicated fields are mandatory.)