
Immunogenicity Evaluation of Biopharmaceuticals
Steven Bowen, PhD, Principal Consultant
The generation of unwanted immune responses to biopharmaceutical products represents a significant risk to product efficacy and patient safety. For this reason, the assessment of product immunogenicity during clinical development is critical. For protein therapeutics, immunogenicity is typically monitored in the form of anti-drug antibodies (ADA) which can be detected in patient serum after exposure to the product. Different products and patient populations carry different levels of risk for inducing an immune response, and assays for immunogenicity testing generally need to be custom designed and validated for each situation. Here we summarize some general considerations for immunogenicity risk assessment, as well as immunogenicity assay development and validation.
Immunogenicity Risk Assessment
The strategy for monitoring ADAs in clinical studies should be informed by the immunogenicity risk of the product, which can be determined from two main questions:
- What is the likelihood that exposure to the product will generate an immune response resulting in ADA?
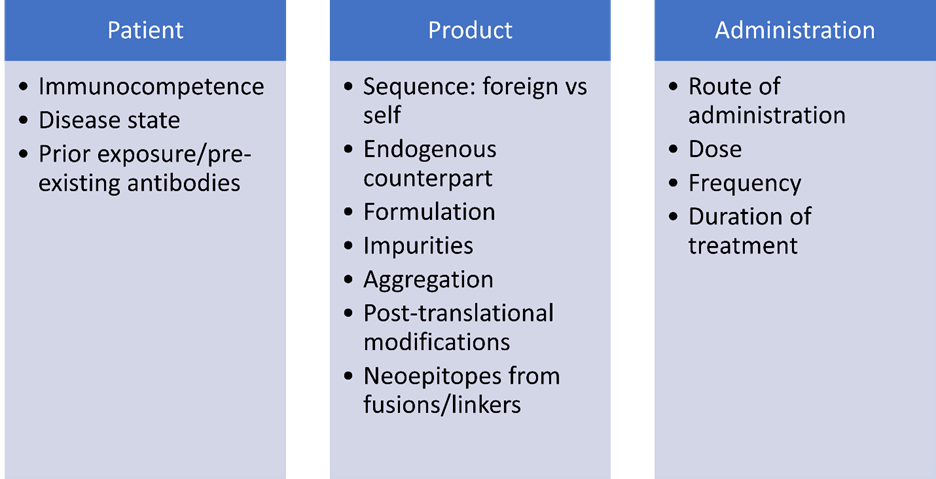
- This will depend on a number of factors, including the amino acid sequence of the molecule (similarity to self proteins), aggregation, post-translational modifications, impurities or other factors in the formulation that could act as adjuvants, the immunocompetence of the patient population, the dose and frequency of administration, and prior exposure to the product which may result in pre-existing antibodies.
- If an immune response that results in ADA does occur, how severe will the consequences be to the patient?
ADA are generally evaluated based on their capacity to 1) bind to the product and 2) interfere with the function of the product. Many ADA have the capacity to bind to the drug, without necessarily impacting its function or pharmacokinetics. A subset of ADA, however, can impact clinical performance by interfering with the drug’s mechanism of action (referred to as neutralizing antibodies or NAbs). Neutralizing antibody responses can have negative consequences for patients including loss of efficacy, and in rare cases, cross-reactivity of NAbs with endogenous proteins that can result in deficiency syndromes. Risk evaluation should consider how severe the consequences of a loss of product efficacy would be for patients. Additionally, if the product has an endogenous counterpart or shares significant sequence homology with a self protein, the risk of the consequences of cross reactivity should be assessed, i.e., if a NAb response is generated against the product, what would happen if those antibodies cross-react with endogenous proteins? Neutralization of endogenous proteins that serve a critical biological purpose and have no redundant function with other proteins would pose the highest immunogenicity risk.
Factors Affecting Immunogenic Potential
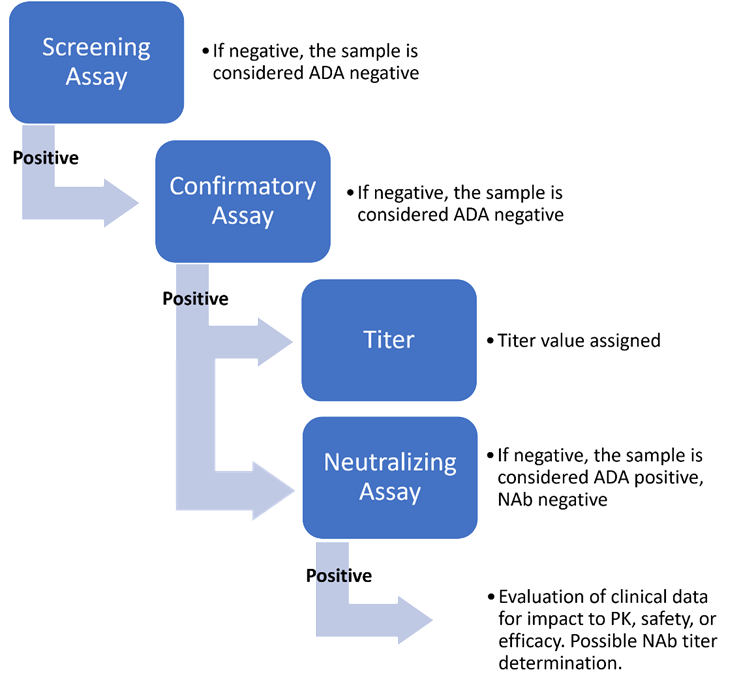
Evaluating Immunogenicity: A Tiered Approach
FDA guidance recommends using a tiered strategy to evaluate ADA in samples collected at appropriate timepoints during clinical studies. This strategy consists of an initial screening assay used on all samples to identify potential binding antibodies, followed by a confirmatory assay performed on all samples that tested positive in the screening assay. Samples that are confirmed as being positive for binding antibodies in the confirmatory assay are generally subjected to a titering assay and a NAb assay. The NAb assay should reflect the product’s mechanism of action and is generally based on the potency assay.
Product-Specific Assay Design
For most products, ADA assays designed to capture ADA against the entire product molecule will be sufficient. However, certain products may require a modified approach to ADA testing that allows for the evaluation of ADA against different parts of the molecule. For example, pegylated protein products have the potential to generate ADA against the PEG moiety, the protein, or both. To understand the specificity of the ADA response, it may be necessary to design assays capable of distinguishing between anti-PEG ADA and anti-protein ADA. Similarly, fusion proteins and antibody-drug conjugates may require unique assay designs that should be considered when developing the overall immunogenicity testing strategy.
Considerations for Validation
In general, ADA assays should be fully validated prior to testing samples from pivotal clinical studies. FDA guidance describes the general Agency expectations for immunogenicity assay validation. However, adequate validation of product-specific immunogenicity assays has remained a challenge for many companies, which can result in extensive information requests during regulatory review and potential delays in approval.
Some critical areas of focus during assay validation include:
- Positive and negative control selection and qualification
- Cut-point determination with relevant serum samples
- Setting of appropriate positive control concentrations for system suitability
- Sensitivity
- On-board drug tolerance of the assay compared to levels expected in patient serum at the time of sampling
- Precision
- Reproducibility
- Accuracy
- Specificity
- Selectivity
- Matrix interference from other substances in the serum, including hemoglobin, lipids and bilirubin
- Determination of the minimum required dilution (MRD) of serum samples prior to analysis
- Robustness/sample stability
The assay validation exercise should be designed to thoroughly address all of the above validation parameters to ensure that the methods used to test patient samples provide reliable and accurate clinical data.
Integrated Summary of Immunogenicity
An integrated summary of immunogenicity (ISI) should be included in the BLA, Section 5.3.5.3 Reports and Analysis of Data from More than One Study. This summary represents a centralized location for the presentation and discussion of immunogenicity data to support the application. The summary should generally include:
- Immunogenicity risk assessment
- Immunogenicity testing strategy at each stage of clinical development
- Clinical study design and sampling strategy as it relates to immunogenicity samples
- Clinical immunogenicity data analysis and discussion
- Conclusions and risk evaluation and mitigation strategy (REMS), if needed
Throughout product development, the assessment of immunogenicity risk and the methods for evaluating product immunogenicity will evolve.
Immunogenicity Assessment for Biosimilar Development
A conclusion of biosimilarity requires that the biosimilar product be “highly similar to the reference product notwithstanding minor differences in clinically inactive components” and that “there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product.” The demonstration of “no clinically meaningful differences” is typically achieved through comparative clinical studies using the proposed biosimilar and the reference product, including a comparative immunogenicity assessment. Inadequate immunogenicity testing can cause significant regulatory challenges for biosimilar product development. Additionally, differing rates of binding ADA or NAbs between a biosimilar and a reference product can raise concerns about the conclusion of biosimilarity.
How ELIQUENT Can Help
Although we have provided a brief introduction above regarding immunogenicity of biologic drugs, there are many complexities that ELIQUENT experts can help sponsors navigate during clinical development including risk assessment, assay design and validation, and interpretation of clinical data. Our immunogenicity experts can provide guidance on critical Agency interactions (e.g., Type B, Type C, BIAM, BPD2, and BPD3 meetings) throughout development where Agency feedback on the immunogenicity testing plan is needed. ELIQUENT can also provide useful strategic advice on the design and validation of immunogenicity assays to ensure that the data will meet Agency expectations.
Ready for regulatory clarity? Contact Us.
(* Indicated fields are mandatory.)