
Not the Usual Suspects – FDA’s Drug Recalls – Oral Dosage Forms Edition
Executive Summary
Recalls are the ultimate reactive measure, and it is a lose-lose situation for all parties involved. In addition to patient impact, a recall is costly both financially and reputationally and may not preclude United States Food and Drug Administration (FDA) regulatory action. FDA’s most recent Report on the State of Pharmaceutical Quality captures recall trends from FY2020 through FY2024. In this white paper, we delve deeper, evaluating recalls specific to oral dosage forms from June 2012 to August 2025. Our focus is not on recalls that are the most frequent, but ones that can happen readily if you are not vigilant. For each recall “unusual suspect” we identify the defect and/or sub-defect group, discuss regulatory requirements, and outline FDA guidance recommendations and other best practices. Our aim is to help prevent similar issues from occurring at drug manufacturing facilities, keep products on the market, and protect patient health.
Methods
A dataset was pulled from FDA’s Data Dashboard for drug recalls from June 2012 to August 2025. This dataset includes recall by line item, whereby different products may be associated with one recall. We recommend a database enhancement to FDA’s recall data, whereby product type is included to enhance regulatory intelligence.
The dataset was filtered to include oral dosage forms using keywords (e.g., oral, tablets, capsules, liquids, syrups, suspensions). Data was filtered to exclude dietary supplements using keywords in product description (e.g., herbal, supplement, CBD, kratom, hemp, sexual) and/or recalling firm names associated with dietary supplements. This dataset was analyzed using a combination of “off-the-shelf” artificial intelligence and human review. The dataset of oral dosage forms was then sorted into seven defect groups. Sub-defect groups included in this white paper were identified using a similar keyword search. These data were then expressed as a bubble chart.
Results
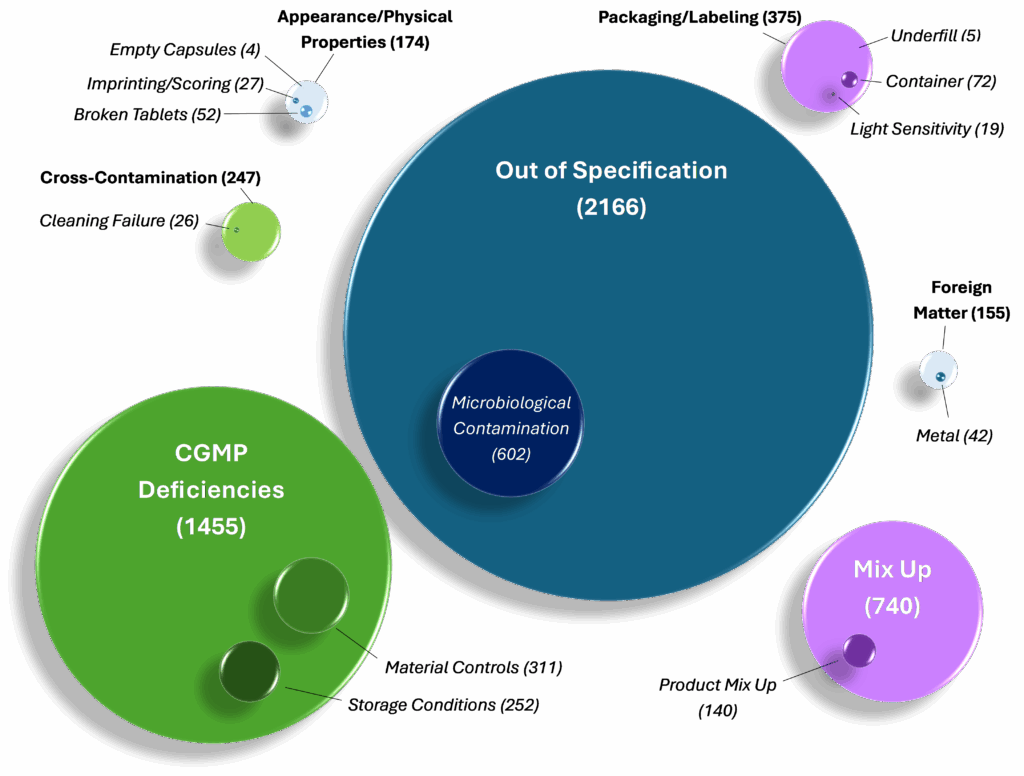
Figure 1. Not the Unusual Suspects – Oral Dosage Form Recalls from 2012-2025. The above figure displays all 5,312 oral dosage form recalls included in FDA’s Data Dashboard from June 2012 to August 2025. These recalls were sorted into the following seven Defect categories (see parent bubbles, above): Out of Specification, CGMP Deficiencies, Mix Up, Cross-Contamination, Appearance/Physical Properties, Packaging/Labeling, and Foreign Matter. A subset of recalls were sorted into Sub-defect categories (see child bubbles, above): Cleaning Validation, Raw Material Controls, Metal, Imprinting/Scoring, Empty Capsules, Broken Tablets, Product Mix-ups, Container Defect, Light Sensitivity, Underfill, Microbiological Contamination, and Storage Conditions. The numbers adjacent each Defect and Sub-defect category correspond to an individual recall line item. Each Sub-defect in the above figure is further elaborated on in the narrative of this white paper.
Discussion
Discussed here is a subset of defect and sub-defect groups that may lead to a drug recall. Most of the recalls in this white paper are due to Current Good Manufacturing Practice (CGMP) deficiencies. When a drug product is manufactured out of full compliance with CGMPs it is considered adulterated per Section 501(a)(2)(B) of the Federal Food Drug and Cosmetic Act. A drug product can also be recalled for misbranding under 502(a)(1) if its labeling is false or misleading.
The Code of Federal Regulations (CFR) references for each defect and sub-defect group discussed in this paper are examples of primary regulations that are likely in violation. However, recalls also involve a concomitant failure of the quality system, and, as such, other quality system regulations may apply. Most notably:
- 21 CFR 211.22 requires the quality unit to ensure only compliant drug products are released. Both the quality unit of the manufacturing facility and the applicant bear responsibility for ensuring the compliance of their drug products on the market.
- 21 CFR 211.100 requires the manufacturing process to be validated to ensure drug products meet specification. Quality is something that must be built in – testing alone is not enough.
- 21 CFR 211.192 requires investigations into any unexplained discrepancies or the failure of a batch or any of its components to meet any of its specification. These investigations must be thorough and extend to other batches regardless of whether they have been distributed. Investigations should include the conclusions and follow-up, namely, root cause identification and effective corrective and preventive actions to prevent recurrence.
Defect: Cross-Contamination, Sub-defect: Cleaning Failure
Hazards that can increase risk of cross-contamination in oral dosage form production include multi-purpose facilities, highly potent products, active pharmaceutical ingredients made with recycled solvents, products that are poorly soluble, dust generation, new product introductions, facility flows, air handling systems, manual cleaning processes, automated cleaning processes, and parts of equipment that are not disassembled in between product changeovers for cleaning.
The federal regulations require that equipment and utensils be cleaned, maintained, and sanitized at appropriate intervals to prevent malfunctions or contamination that would alter the safety, identity, strength, quality, or purity of the drug product.
Prior to manufacturing, equipment must be clean. Cleaning procedures should consider and mitigate risks such as the variability of hand scrubbing and the residues after solvent rinses. Procedures should clearly address equipment cleaning for water soluble and non-water-soluble residues as appropriate. Equipment that is difficult to clean may be best addressed via use of dedicated equipment. Dedicated equipment; however, does not mitigate the risk associated with accumulation of product-related impurities.
Cleaning validation protocols should identify “worst case” sample locations and include swab tests, total organic carbon (TOC) measurements, and rinse samples. Methods should be both validated for detection limits and proven for recovery and specificity. Residue limits should relate to therapeutic dose or health-based limits for the US or EU market, respectively. Maximum allowable carryover (MACO) calculation errors can happen, and calculations should be verified.
These considerations should also cover the contracts between applicants and contract manufacturing facilities. Applicants should request notification of the addition of any new molecular entity added into the contract facility that will be manufactured on shared equipment. For cleaning following investigations of cleaning failures, carefully consider the scope of the investigation as well as how you will ensure the entire equipment train is in fact clean.
Note, the number of cross-contamination failures related to cleaning is likely higher; however, only recalls where cleaning was identified in the recall reason were highlighted.
Defect: CGMP deficiencies, Sub-defect: Material Controls
Raw material quality is critical to ensure finished drug product quality, but it is also a variable outside of your direct control. In order to ensure raw materials remain in a state of control, risk-based assessments should be performed. Sole suppliers should be managed with additional care by, for example, considering redundant suppliers (where possible), stockpiling, and knowing the raw material supplier’s business continuity plans. Paper assessments should not be used in place of on-site audits for high-risk suppliers. Supplier qualification is not a one-time operation; it needs periodic re-examination. Best practice includes having a system in place to detect and respond to changes in materials from suppliers to prevent unintended consequences.
Contracts should be thoughtfully considered and factor in scenarios for when a supplier may provide you with material that does not meet specification. They should include elements to ensure that your complaints are adequately investigated and list who will be responsible for the cost of additional testing. Qualification procedures should also ensure that both qualification status and testing regimen are changed.
Defect: Foreign matter, Sub-defect: Metal
Consider what is required to mitigate the risk of metal particulates in drug product. Equipment should be designed to minimize metal on metal contact that may generate metal particulates. Regular maintenance should be scheduled to verify that equipment is operating appropriately and is visually acceptable. A replacement schedule should be established for parts and should be based on a sound rationale, e.g., manufacturer recommendations and in-house data. Punch tips and other tooling should be on a regular maintenance schedule, and maintenance frequency should factor in drug formulation and frequency of use. Metal detectors should be of appropriate design and be validated, calibrated, and verified to ensure they are sensitive enough to detect low levels of contamination.
Defect: Appearance/Physical properties, Sub-defect: Imprinting/Scoring
21 CFR 206, 21 CFR 201.57, 21 CFR 211.67
Tablet printing and imprinting is critical to ensuring appropriate identification of the tablet. Tablet scoring is especially critical for medications requiring precise dose titration, such as steroids and hypertensives. Setup operations, tablet attributes (including shape dimensions and surface characteristics), and punch wear, automated visual inspection, and reject segregation can play a role in the ability to print, imprint, or score a tablet.
Defect: Appearance/Physical Properties, Sub-defect: Empty Capsules
Empty capsules are a serious issue, especially if the medication in question is needed to keep a patient from hospitalization. Empty capsules could occur because they do not separate during manufacturing. As with any component, supplied capsules should be tested against the certificate of analysis (including for dimensional specifications) on a periodic basis. Capsules have sensitive storage requirements and must not be stored outside the recommended ranges for temperature or humidity. Equipment needs to be maintained to ensure appropriate separation of capsules during filling operations. Lines should be equipped with an empty capsule eliminator, and this equipment should be verified for appropriate operation.
Defect: Appearance/Physical Properties, Sub-defect: Broken Tablets
21 CFR 211.84, 21 CFR 211.100(a), 21 CFR 211.192, 21 CFR 211.46
Tablets, regardless of their disintegration rate requirements, must withstand the stresses from manufacturing, distribution, and dispensing. It is first important to qualify what type of break is occurring (e.g., capping, chipping, cracking) to determine what root causes may be at play. Additional testing beyond tablet thickness, breaking force, and friability may be necessary to determine the issue. Root causes can range from materials (excipient type or content, powder density), equipment (equipment maintenance and tooling condition), environment (e.g., humidity), or process variability (mixing time, tablet thickness, coating, manufacturing deviations). Best practice includes ensuring that the formulation is right from the outset, understanding the role and vulnerabilities of components, and building in robustness where possible. A lifecycle approach, from research and development to knowledge management, is critical to ensure the carryover of process knowledge.
Defect: Mix-ups, Sub-defect: Product Mix-ups
While mix-ups are only mentioned four times in 21 CFR 211 (and only once in FDA’s Compliance Program Guidance Manual used for inspections of oral dosage manufacturers), mix-ups are a critical issue and must be adequately controlled. Mix-ups can occur at the labeling or the product (dosage form) level. Labeling mix-ups are the most common of the two. Product mix-ups for oral solid dosage forms can occur at tableting/encapsulation, bottling/blistering, or dispensing. Mix-ups can even occur within a product batch if inspection systems and waste streams are not properly controlled. It is important to have a firm understanding of what products are manufactured at your site or your contract site, the equipment used, and the maintenance, cleaning, line clearance, and set up procedures/practices in order to fully understand mix-up risk as well as to perform adequate investigations into any complaints of mix-ups. Risk can be mitigated via use of “entrapment free” equipment; however, it is important to note that this equipment facilitates—but does not replace—maintenance, cleaning, line clearance, and set up operations.
Defect: Packaging/Labeling, Sub-defect: Container
Regulations for supplier controls referenced earlier for raw materials also apply to drug product containers and closures. Recalls have occurred in cases where product containers leak. Best practice should ensure that containers are tested periodically—not only at receipt—and on stability in various orientations. Packaging risks may also extend to co-packaged containers including dosing cups, which should be examined and tested to ensure conformance to specifications.
Defect: Packaging/Labeling, Sub-defect: Light Sensitivity
Consider the case of a recall for a drug that is not repackaged in accordance with the requirements on the original label. Regulations require drug products to be tested for stability in the same container-closure system in which the drug product is marketed. Drug products may be repackaged into unit doses for ease of dispensing under healthcare settings; however, this practice must ensure that stability of the product is not compromised. If the assigned expiration dates do not exceed 6 months from date of repackaging or 25 percent of the time between the date of repackaging and the original expiration, FDA does not intend to take action regarding required stability studies for repackaged solid oral dosage forms provided that the following conditions are met: (1) the container complies with USP Class A or B standards as described in General Chapter <671>, (2) the container protects light sensitive products where applicable, (3) the container is opened and repackaged in one operation, (4) the container has storage conditions that comply with the original drug product label or USP <659>, and (5) the original drug product label does not caution against repackaging. Should expiration not be reduced per guidance, data from appropriate studies are required to demonstrate that product quality is maintained until the expiration date.
Defect: Packaging & Labeling, Sub-defect: Underfill
Underfilled units can be a serious issue for public health, particularly when a patient is unable to get their prescribed dose of medication. Consider a case where 30ct bottles are short by 1-3 tablets. In this case, weight checks on the filling line can help prevent this issue from occurring, but it is not that simple. Where the weight-checker is placed, its sensitivity, maintenance, and calibration are all critical to ensure dosage counts are accurate. One technology that can add greater control is an electric eye counter, but this also requires appropriate qualification in order to ensure adequate functionality.
Defect: Out of Specification (OOS), Sub-defect: Microbiological contamination
21 CFR 211.22, 21 CFR 211.67, 21 CFR 211.84(d)(4)
Out of specification results are a large reason for drug recalls. Drug products must meet requirements for identity, strength, quality, and purity. There are many points of failure for an oral dosage form, including dissolution, potency, nitrosamine and azido impurities, and chemical impurities. The quality unit is responsible for ensuring its drug products meet these requirements. One area that may be overlooked is the microbiological requirements for oral dosage forms. While most oral dosage recalls for microbiological contamination relate to liquid dosage forms known to promote growth of microorganisms, there are also recalls for oral solid dosage forms.
The United States Pharmacopeia (USP) chapters for nonsterile products, <60> Microbiological Examination of Nonsterile Products Tests of Burkholderia Cepacia Complex, <61> Microbial Enumeration Tests and <62> Tests for Specified Microorganisms, identify the limits of microorganisms in oral dosage forms as well as tests performed to ensure certain objectionable microorganisms are absent. FDA drug recalls for oral dosage forms have also occurred for other microorganisms not listed in this chapter, including B. cereus. USP includes informational chapters such as <1111> Microbiological Examination of Nonsterile Products and <1115> Bioburden Control of Nonsterile Drug Substances and Products to help determine whether or not a microorganism can be considered objectionable. Additionally, draft FDA guidance is available to help establish appropriate specifications and manufacturing controls to prevent drug product contamination with microorganisms.
Defect: CGMP Deficiencies, Sub-defect: Storage Conditions
21 CFR 211.142, 21 CFR 211.150
Regulations require storage of drug products under appropriate conditions of temperature, humidity, and light so that the identity, strength, quality, and purity of drug products are not affected. Once a drug has left your facility, the risks do not end. Recalls have occurred in the distribution space due to temperature abuse. While the error may ultimately lie with your distributor, consider the impact on the patient who cannot get their medication and the impact to your reputation.
Conclusion
Drug recalls can be prevented through proactive quality oversight to prevent problems before they occur. A recall may prevent further patient harm by removing non-compliant product on the market, but it may also create a drug shortage that ultimately still harms the patient who is unable to get their treatment. Ultimately, an effective pharmaceutical quality system and effective risk management, as described in FDA guidance, are foundational to ensuring that your company can meet the needs of patients, health care professionals, and regulatory authorities.
How ELIQUENT Can Help
Although we have provided a brief summary regarding select recall defect groups and sub-groups among oral dosage forms, there are additional drug dosage forms, regulatory concerns, and complexities that may pose a compliance risk if not appropriately mitigated. ELIQUENT experts can help drug manufacturing facilitiess navigate these concerns through on site or remote assessments. Our Regulatory Compliance team can provide you with appropriate guidance to ensure your drug products meet FDA expectations.
References
- Federal Food, Drug, and Cosmetic Act
- 21 CFR 211
- Validation of Cleaning Processes (7/93)
- Dosage Form Drug Manufacturers cGMPs (10/93)
- Guidance for Industry Quality Systems Approach to Pharmaceutical CGMP Regulation
- Contract Manufacturing Arrangements for Drugs: Quality Agreements
- Expiration Dating of Unit-Dose Repackaged Solid Oral Dosage Form Drug Products
- CPGM 7356.002 Drug Manufacturing Inspections
- United States Pharmacopeia and National Formulary
- Guidance for Industry Process Validation: General Principles and Practices, January 2011, Revision 1
- Guidance for Industry Q10 Pharmaceutical Quality System, April 2009
- Guidance for Industry Q9(R1) Quality Risk Management
- Draft Guidance for Industry Microbiological Quality Considerations in Non-sterile Drug Manufacturing, September 2011
Acknowledgments
The authors acknowledge ELIQUENT Principal David Elder for his review, Laura Bartee for her amazing editorial skills, and Kalah Auchincloss for her support.
Ready for regulatory clarity? Contact Us.
(* Indicated fields are mandatory.)