
Transitioning to E2B(R3) Electronic Safety Reporting for INDs
Key Takeaways:
- E2B(R3) is the new global standard for electronic safety reporting of IND-associated ICSRs, replacing legacy formats with structured XML reporting.
- Sponsors must update safety reporting systems and workflows to meet FDA IND safety reporting requirements before the April 2026 compliance deadline.
- Two submission pathways exist: direct database-to-database transmission via the FDA Electronic Submissions Gateway (ESG) or manual entry through the FDA Safety Reporting Portal (SRP).
- Not all safety reports qualify for E2B(R3); certain IND safety reports from preclinical studies or other sources still require eCTD submission due to narrative and contextual needs.
- Compliant ICSR pharmacovigilance demands accurate routing identifiers, schema validation, and adherence to FDA business rules to avoid rejections.
- Transition impacts multiple operational areas, including regulatory submission automation, traceability with automated acknowledgements (ACK1/ACK2), and pharmacovigilance data management.
Pharmacovigilance plays a critical role in safeguarding public health by continuously monitoring the safety and efficacy of pharmaceutical products throughout their lifecycle. The prompt and accurate reporting of patient-level adverse events to regulatory authorities is essential to identify potential safety signals, assess risks, and implement appropriate risk-mitigation measures to protect patient safety. An individual case safety report (ICSR) is an integrated report captures information describing adverse event(s) / reaction(s) experienced by an individual patient associated with the use of FDA regulated products[1]. These reports need to be transmitted across health authorities, throughout the health product lifecycle, from early clinical investigation to post-marketing surveillance. As ICSR pharmacovigilance volumes grow, efficient and compliant reporting becomes increasingly essential. Over the last decade as the number of case reports has increased, reporting of ICSRs has evolved from paper-based submissions to standardized electronic reporting.
Given the high volume and complexity of worldwide safety data exchange, there has been a critical need for a harmonized data standard that allows reports to be generated, transmitted, and processed automatically across different systems.
To meet this need, International Council for Harmonisation (ICH) developed what is referred to as the E2B(R3) data standard that defines the format, structure, and content of ICSRs transmitted electronically to regulatory agencies. As a member of ICH, the US FDA has implemented the E2B(R3) standard as part of its modernization of FDA IND safety reporting systems, requiring sponsors to transition from legacy formats to the structured E2B(R3) framework for investigational before April 1, 2026. This article is intended to help sponsors understand what the change entails, how the implementation process works, and what practical steps are needed to ensure a smooth and compliant migration for IND safety reporting requirements.
[1] 21 CFR 310.305(b), 314.80(a), and 600.80(a); see also 21 CFR 329.100(b).
Under the new process, there are two options for submitting the documents (Figure 1):
- ICSR Option A: Database-to-Database Transmission – Submitters who have database-to-database transmission capability may directly submit ICSRs in the XML format via the FDA Electronic Submissions Gateway (FDA ESG)
- ICSR Option B: Safety Reporting Portal (SRP) – Requires registration and receipt of login credentials – Submitters enter the ICSR information manually into a web-based form and submit – Submitted ICSR uploaded into the FAERS database. This option is aligned with the FDA Safety Reporting Portal used for manual entry submissions.
Figure 1. Submission Pathways for Safety Reports Under FDA’s Implementation of The ICH E2B(R3) Standard
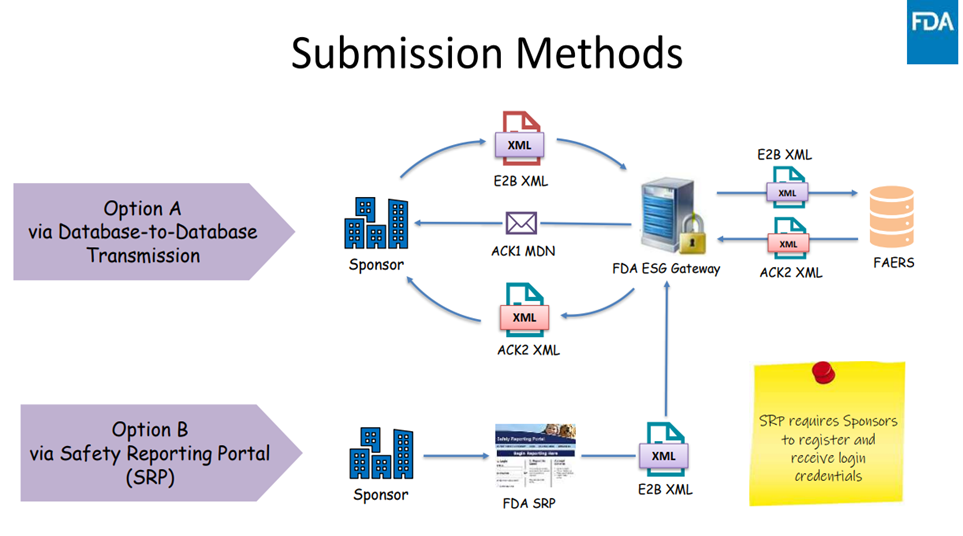
Please note, not all IND safety reports will go to FAERs. As detailed in Table 1, certain types of IND safety reports are not submitted using the E2B(R3) data structure. Reports such as findings from other studies, animal or in-vitro testing results, or observations of increased incidence rates are reports that summarize aggregated or preclinical data. These reports require contextual narrative explanation, study interpretation, and supporting documentation, therefore, they cannot be captured within the standardized ICSR XML fields and still need to be submitted in Electronic Common Technical Document (eCTD) format. This remains a key consideration in clinical trial safety reporting and regulatory submissions.
Table 1. Submission Pathways for Different Types of IND Safety Reports Under 21 CFR 312.32
| Type of IND Safety Report | Submit to FAERS | Submit in eCTD Format |
| A single occurrence of an event that is uncommon and known to be strongly associated with drug exposure (21 CFR 312.32(c)(1)(i)(A)) | X | |
| One or more occurrences of an event that is not commonly associated with drug exposure, but is otherwise uncommon in the population exposed to the drug (21 CFR 312.32(c)(1)(i)(B)) | X | |
| An aggregate analysis of specific events observed in a clinical trial (known consequences of the underlying disease or condition) that indicates those events occur more frequently in the drug treatment group than in a concurrent or historical control group (21 CFR 312.32(c)(1)(i)(C)) | X | |
| Findings from other studies (21 CFR 312.32(c)(1)(ii)) | X | |
| Findings from animal or in vitro testing (21 CFR 312.32(c)(1)(iii)) | X | |
| Increased rate of occurrence of serious suspected adverse reactions (21 CFR 312.32(c)(1)(iv)) | X |
In the FDA’s implementation of the E2B(R3) standard, every ICSR transmitted through the ESG requires routing identifiers to ensure proper delivery to the correct receiving division and database. The Batch Receiver Identifier (N.1.4) identifies the overarching destination, while the Message Receiver Identifier (N.2.r.3) specifies the precise regulatory center and report type. For premarket safety reports, the header attributes and routing IDs are separated from post-market pathway, between CDER and CBER and among different report types. Rejection would occur if the submission was not assigned to the correct pathway. The details of the identifiers are listed in Table 2.
Table 2. Batch Receiver Identifier (N.1.4) and Message Receiver Identifier (N.2.r.3)
| Submission Type | AS2 Header | Routing ID | N.1.4 Value | N.2.r.3 Value |
| Premarket | CDER IND ICSR Destination: “CDER” XML Files: AERS_PREMKT_CDER | XML Files: FDA_AERS_PREMKT_CDER | ZZFDA_PREMKT | CDER_IND |
| CBER IND ICSR Destination: “CBER” XML Files: AERS_PREMKT_CBER | XML Files: FDA_AERS_PREMKT_CBER | ZZFDA_PREMKT | CBER_IND | |
| CDER IND-exempt BA/BE ICSR Destination: “CDER” XML Files: AERS_PREMKT_CDER | XML Files: FDA_AERS_PREMKT_CDER | ZZFDA_PREMKT | CDER_IND_EXEMPT_BA_BE | |
| Post-market | Post-market ICSR Destination: “CDER” XML Files: AERS | XML Files: FDA_AERS | ZZFDA | CDER |
In short, the transition modernizes the process — but requires sponsors to align their safety systems, data management, and vendor coordination accordingly. Many operational challenges faced by industry today relate to scaling pharmacovigilance systems to meet increasing pharmacovigilance reporting requirements.
The Transition Process — How ELIQUENT Can Help
ELIQUENT recommends proceeding with Option A of the ICSR submission, in which the Sponsor/CRO establishes a direct database-to-database transmission method with the FDA through the FDA ESG. Our team can support this transition and facilitate communication between the relevant technical groups as needed.
As an alternative, ELIQUENT can submit the XML files provided by the Sponsor/CRO and manage the associated tracking file. Under this approach, we will transmit the reports through the ESG gateway and return the two FDA acknowledgements (ACKs) back to the Sponsor for archiving. The workflow is summarized below in Figure 2.
Figure 2. Process Flow When ELIQUENT Conducts ICSR Submissions on Behalf of the Sponsor

- File Ownership: Sponsor/CRO authors and validates XMLs and ELIQUENT transmits the XMLs to ESG.
- Validation: ELIQUENT confirms each XML is validated through the FDA Validator before transmission.
- Secure Handoff: Sponsor/CRO and ELIQUENT mutually agree on delivery methods and turnaround times.
- Audit Trail: ELIQUENT maintains logs, validation reports, and ACK files as part of regulatory documentation and shares with the Sponsor after completion of each submission.
ELIQUENT offers comprehensive support for sponsors navigating this change. We can serve as the FDA-authorized transmitter via ESG on behalf of the sponsor. We will manage the E2B(R3) test and production submissions to FDA and also maintain secure receipts, transmissions, and acknowledgment tracking. ELIQUENT can act as both the transition advisor and operational transmitter to ensure seamless compliance, optimized regulatory submission automation, and effective management of IND safety reporting requirements and ICSR reporting processes.
Meta Title: Transitioning to E2B(R3): A Guide to IND Safety Reporting Compliance
Meta Description: Understand FDA E2B(R3) IND safety reporting requirements and learn how to implement compliant ICSR submissions.